Next: Example Refinement Up: Determination of Standard Values Previous: Determination of Standard Values
![]()
![]()
![]()
Next: Example
Refinement Up: Determination
of Standard Values Previous: Determination
of Standard Values
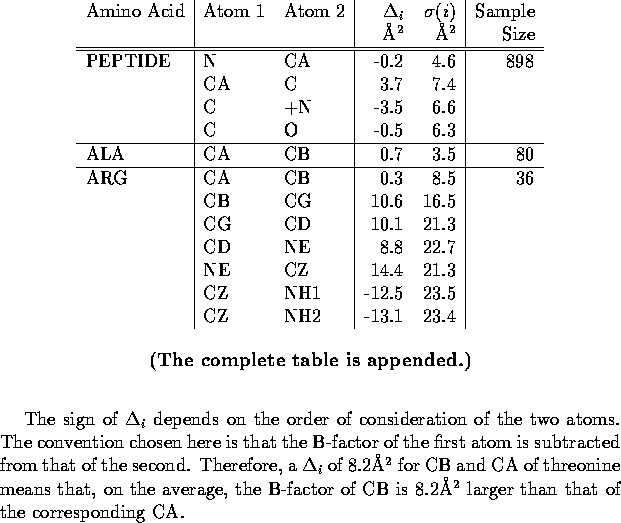
Table 2 summarizes the analysis
of the B-factors for the peptide group, the 20 amino acids, and the disulfide
bond. As anticipated, adjacent atoms within peptide groups have, on average,
fairly similar B-factors. In contrast, a number of the amino acids display
substantial increases in their B-factors as one proceeds from the ![]() -carbon toward the more distal atoms.
-carbon toward the more distal atoms.
As a specific example, consider threonine and valine. These amino acids
are isostructural but have very different hydrophobicity and occur in very
different chemical environments. In threonine the B-factor of the C ![]() atom is 8Å
atom is 8Å ![]() larger than that of C
larger than that of C ![]() while in valine the corresponding difference is only 4Å
while in valine the corresponding difference is only 4Å ![]() . This observation is consistent with valine's preference for being located
in the well ordered core of the protein. In addition, the standard deviations,
. This observation is consistent with valine's preference for being located
in the well ordered core of the protein. In addition, the standard deviations,
![]() , are larger for the restraints in threonine because there is more variability
in the location of these side chains. Threonine residues are sometimes
buried (and ordered) and other times solvent exposed (and disordered) while
valine residues are almost always internal (and well ordered). The smaller
standard deviation for valine allows the B-factor correlation restraint
to be held more tightly in this case because one has greater confidence
in the validity of the restraint.
, are larger for the restraints in threonine because there is more variability
in the location of these side chains. Threonine residues are sometimes
buried (and ordered) and other times solvent exposed (and disordered) while
valine residues are almost always internal (and well ordered). The smaller
standard deviation for valine allows the B-factor correlation restraint
to be held more tightly in this case because one has greater confidence
in the validity of the restraint.
In addition to the library itself, one must determine how closely new
models will be forced to agree with these restraints. In other words, what
is the target value for the final root-mean-square deviation from the ideal
values. The target value can be estimated using cross validation. In cross
validation the statistical analysis is repeated with a subset of data excluded
and the agreement between the subset and the test analysis is monitored.
In this case a new library of ![]() and
and ![]() restraints is determined with one protein excluded and the root-mean-square
deviation of the bonds in the excluded protein compared to the test library
is calculated. This calculation is repeated with each protein excluded
in turn, resulting in ``complete'' cross validation. This results of this
test are listed in Table 3.
In refinement protein models are expected to agree with this library with
a root-mean-square error of 6.5Å
restraints is determined with one protein excluded and the root-mean-square
deviation of the bonds in the excluded protein compared to the test library
is calculated. This calculation is repeated with each protein excluded
in turn, resulting in ``complete'' cross validation. This results of this
test are listed in Table 3.
In refinement protein models are expected to agree with this library with
a root-mean-square error of 6.5Å ![]() .
.
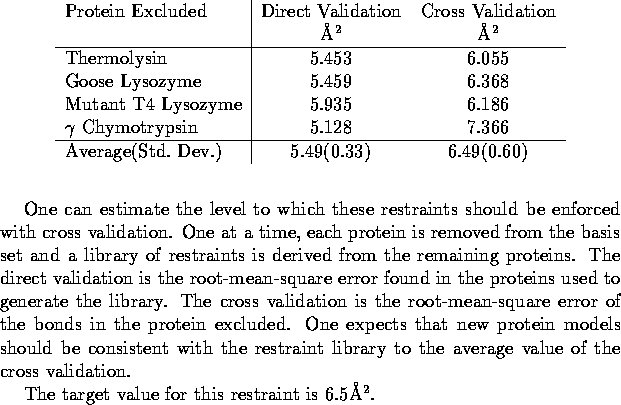
Table 3: Estimation of Target Value