The first extensive use of the package was in the refinement of the bacteriochlorophyll a protein (Tronrud, Schmid & Matthews, 1986). This molecule (molecular weight 150 000 daltons) consists of three identical subunits related by a threefold axis of symmetry. Each of the subunits consists of a polypeptide chain of approximately 350 amino acids that enclose seven bacteriochlorophyll a molecules (Fenna & Matthews, 1975; Matthews, Fenna, Bolognesi, Schmid & Olson, 1979). During the course of the refinement the amino-acid sequence of the protein was not known, but has been reported subsequently (Daurat-Larroque, Brew & Fenna, 1986).
There were several factors that led to the adoption of the present refinement package. The first was the size of the computation problem. As summarized in Table 4,
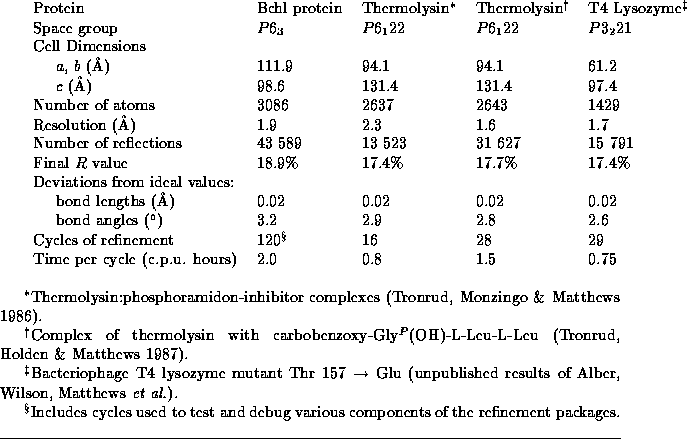
Table: Representative refinements of macromolecular structures
the asymmetric unit contains 3086 atoms and there are 43 598 reflections to 1.9Å
resolution. In addition, the space group ( ![]() ) precludes full use of the crystallographic
symmetry to reduce the size of the FFT calculations. Experience with other projects
in the laboratory suggests that the Hendrickson refinement program (Hendrickson & Konnert, 1980)
would require about 48h of c.p.u. time per refinement cycle on our VAX 11/780
(subsequent improvements to the Hendrickson program have substantially improved
its computational efficiency). We had also had experience in the laboratory with
EREF (Jack & Levitt, 1978). This program is substantially faster, on a
per cycle basis, than the Hendrickson program. A disadvantage of using EREF
for the refinement of the Bchl protein arose from the presence of the Bchl rings.
Because of the diversity of bond lengths and bond angles in the seven bacteriochlorophylls,
as well as uncertainties in their energetics, the definition of standard geometry
for use by EREF appeared to be quite difficult.
) precludes full use of the crystallographic
symmetry to reduce the size of the FFT calculations. Experience with other projects
in the laboratory suggests that the Hendrickson refinement program (Hendrickson & Konnert, 1980)
would require about 48h of c.p.u. time per refinement cycle on our VAX 11/780
(subsequent improvements to the Hendrickson program have substantially improved
its computational efficiency). We had also had experience in the laboratory with
EREF (Jack & Levitt, 1978). This program is substantially faster, on a
per cycle basis, than the Hendrickson program. A disadvantage of using EREF
for the refinement of the Bchl protein arose from the presence of the Bchl rings.
Because of the diversity of bond lengths and bond angles in the seven bacteriochlorophylls,
as well as uncertainties in their energetics, the definition of standard geometry
for use by EREF appeared to be quite difficult.
As discussed previously, the definition of standard geometry in the present refinement package is very flexible, and readily adaptable to `unusual' situations. In particular, in the present situation it was not necessary to force all the conjugated atoms in the Bchl rings to lie in a single plane. Rather, we divided the conjugated atoms into appropriate sets of overlapping sub-planes (Tronrud, Schmid & Matthews, 1986). This method of restraint maintained local planarity, but allowed larger-scale deformations. This procedure led to the finding that the seven Bchl rings exhibit two distinct classes of bending, one of which is also observed in the structure of ethyl chlorophillide a (Tronrud, Schmid & Matthews, 1986). An inappropriate application of restraints to the Bchl rings could well have masked this small but significant effect.
In the initial refinement of the Bchl protein the refinement package used analytical summations to calculate the gradient of the crystallographic term. With this method each cycle of refinement required 8h of c.p.u. time on our VAX 11/780. The present version of the program requires 2.0h for the same problem.
The general strategy of refinement that we have adopted for the Bchl and other
proteins is first to refine for several cycles with weak geometry restraints,
then to run a few cycles of temperature-factor refinement, then restore the model
to good stereochemistry by refining for several cycles with strong geometry restraints
and finish with several additional cycles of thermal factor refinement. At this
stage the resulting difference electron density map and ` ![]() ' map are
inspected on the graphics system in the usual way. Potential problem areas are
highlighted by inspecting the lists of worst bond lengths, bond angles, departures
from possible errors in the assumed amino-acid sequence. In this instance the
cycles of refinement followed by inspection of the model were repeated seven times
to achieve the final refined structure and `X-ray' amino acid sequence. The
overall refinement statistics are summarized in Table 4.
' map are
inspected on the graphics system in the usual way. Potential problem areas are
highlighted by inspecting the lists of worst bond lengths, bond angles, departures
from possible errors in the assumed amino-acid sequence. In this instance the
cycles of refinement followed by inspection of the model were repeated seven times
to achieve the final refined structure and `X-ray' amino acid sequence. The
overall refinement statistics are summarized in Table 4.